|
DAUMANTAS MATULIS |
Structure and Thermodynamics for Drug Design
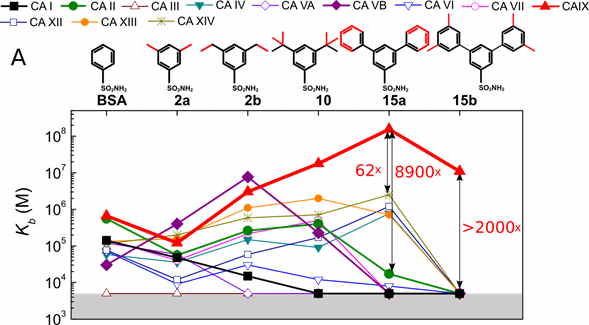
Fig. 1.
Rational drug design attempts to discover a ligand that would bind a disease target protein with high affinity and selectivity over unintended targets to avoid toxicity. Despite significant efforts, the underlying physical forces that determine the protein-ligand recognition are still rather poorly understood.
To help design compounds and predict their affinity to target proteins, we are assembling datasets, where chemical compounds binding to proteins would be characterized, including
(a) the X-ray crystallographic structures of protein-ligand complexes,
(b) the thermodynamics of interaction including the enthalpy, entropy, Gibbs energy, volume, heat capacity and other thermodynamic parameter changes upon binding,
(c) the kinetics of the same protein-ligand binding, including the on- and off-rates.
We are primarily focused on the human family of twelve catalytically active carbonic anhydrase isoforms as a disease protein-target. These enzymes have essentially the same fold and a highly similar shape of the active site suitable for the testing of isoform selectivity.
The group of scientists come from various backgrounds including molecular biologists, biochemists, organic chemists, biophysicists, physicists, computer modellers, biologists and pharmacists. Organic synthesis scientists design and perform the synthesis of novel compounds, molecular and cellular biologists perform the cloning, expression (both in bacterial and in human cell cultures) and purification of target proteins, biothermodynamicists determine the energetics of binding between the synthesized compounds and the target proteins by isothermal titration calorimetry or thermal shift and search for structure-energetics correlations, crystallographers determine the X-ray crystallographic structures of protein-compound complexes, in silico modellers perform compound docking, and the pharmaceutical scientists perform development studies of the effect of compounds in various biological systems including cancer cell cultures, zebrafish and mice.
SELECTED PUBLICATIONS
- Dudutienė, V., Zubrienė, A., Kairys, V., Smirnov, A., Smirnovienė, J., Leitans, J., Kazaks, A., Tars, K., Manakova, L., Gražulis, S., Matulis, D. Isoform-selective enzyme inhibitors by exploring pocket size according to the lock-and-key principle. Biophys. J. 2020, 119(8): 1513–1524.
- Kazokaitė-Adomaitienė, J., Becker, H. M., Smirnovienė, J., Dubois, L. J., Matulis, D. Experimental approaches to identify selective picomolar inhibitors for carbonic anhydrase IX. Current Medicinal Chemistry. 2020. doi: 10.2174/0929867327666201102112841.
- Zakšauskas, A., Čapkauskaitė, E., Jezepčikas, L., Linkuvienė, V., Paketurytė, V., Smirnov, A., Leitans, J., Kazaks, A., Dvinskis, E., Manakova, E. et al. Halogenated and Di-substituted benzenesulfonamides as selective inhibitors of carbonic anhydrase isoforms. European Journal of Medicinal Chemistry. 2020, 185, 111825.
Compound Binding to Proteins via ‘Induced Fit’ or ‘Lock-and-Key’
Compounds bind to proteins via different mechanisms ranging from ‘induced fit’ to ‘lock-and-key’ modes. The induced fit emphasizes the fact that ligands may alter the protein structure and dynamics upon binding. The ligand-bound and ligand-free states of a protein may differ significantly. The lock-and-key principle is applied when the protein is conformationally rigid and does not visually change its structure upon ligand binding. This seems to us to be the case despite variation in the literature and it is important to design ligands that would fit the active site pocket of the disease target protein as closely as possible.
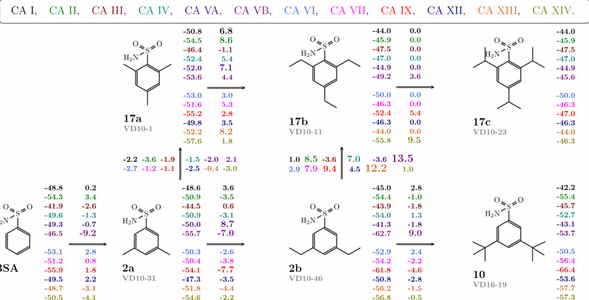
We have designed a series of compounds with enlarged sizes until they could not fit in the active site and the affinity dropped thousand-fold upon addition of a minor substituent such as a methyl group (see Fig. 1). A series with a substituted hydrophobic group of increasingly larger size was used. If the protein is rigid, there ought to be a limiting size after which the compound cannot fit into the binding site. This size limit should vary for every isoform and could be employed to search for isoform-selective inhibitors. Additionally, the size of the ligand would prevent it from binding to isoforms that are of vital importance and should not be targeted.
The map of correlations between the compound chemical structures and the intrinsic standard Gibbs energy changes upon compound binding to all 12 CA isoforms shows the energies next to the chemical structures (see Fig. 2). Differences in the energies upon binding comparing chemically similar compounds are shown next to the arrows connecting the compounds. Colours represent the 12 catalytically active human CA isoforms.
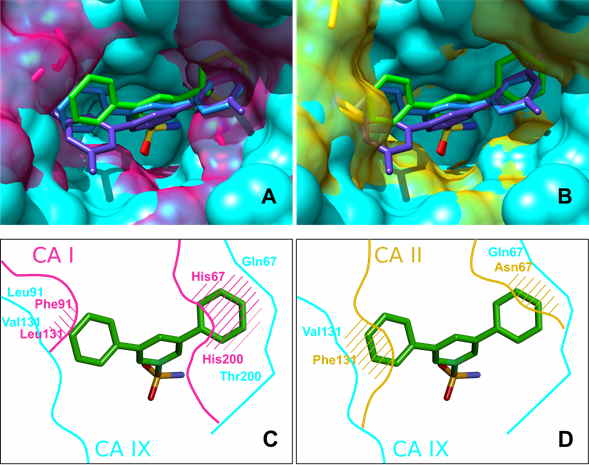
X-ray crystallographic analysis of several compounds bound to CA II, CA IX and CA XII essentially confirmed that the most successful ligands nearly completely filled the binding site. X-ray crystallography confirmed that the selectivity of 15a towards CA IX was due to the optimal size of the compound and the binding site, e.g. due to the decreased size of residue 131, which has changed from Phe in many CA isoforms to Val in CA IX.
Docking of all compounds to all CA isoforms was employed to test the extent to which we could predict the structural positions and the binding energetic (see Fig. 3). The docking calculations were able to correctly reproduce the binding modes of larger ligands, such as 24, 10, and 15a, with mixed success in reproducing the binding modes of the smaller ligands. For the important CA IX and CA XII isoforms, the docking essentially confirmed conclusions drawn from the X-ray structures. Thus, it has a predictive capability that can be used to design better drugs.